
 400-1050-986
400-1050-986 市场资讯
市场资讯新闻资讯 更多+
1、全球发展概况:随着全球医药市场竞争的日益激烈,药企对于研发、生产、销售成本的控制和效率的提升需求强烈催生了医药外包行业的产生。此后,医药外包行业又逐渐分化为专业的研发外包(CRO)、生产外包(CMO)、临床试验现场管理外包(SMO)等。20世纪70年代SMO在美国诞生,通过提供现场管理,帮助临床试验机构搭建临床试验的执行体系。临床试验机构通过借助SMO企业资源在短时间内迅速组织起具有高度专业化和丰富经验的临床试验团队,更加有效地控制研发管理费用及协调内外部资源配置。与国内不同的是,欧美等国家无临床研究机构准入门槛,在有条件的公立医院、私人医院和诊所均可开展临床研究,故SMO参与到大量临床试验中。此后,20世纪90年代SMO在欧美及日本迅速扩张,经过近三十年的发展,SMO逐渐成为医药研发产业链中不可缺少的环节。
SMO拥有较为丰富的临床试验现场管理经验,能为临床试验机构节省大量资料统计、数据誊写等繁杂工作,使研究者能够将精力集中于临床研究的医学判断,为临床研究质量提供了保证。随着临床试验机构管理工作的日益细化,由专业的临床研究协调员参与临床试验并做好现场管理协调工作将是未来的趋势,临床试验机构也逐渐接受使用第三方临床研究协调员的工作模式。
2、我国SMO发展概况:
(1)SMO发展历程:随着临床研究的全球化,以及中国在病人资源和运营成本等方面的优势,越来越多的国际多中心临床研究进入中国,中国在世界新药研发及临床研究中的地位愈发重要。
在此背景下,2008年行业内开始出现模仿国外SMO开展部分业务的企业,建立了以人力派遣为主的CRC团队参与试验,并在报价、管理、培训等商业模式上搭建了中国SMO的基本框架。2005年3月1日起,我国药监部门联合卫生部要求对临床试验机构实行资格认定制,未通过认证的机构将不再具有承担药物临床试验的资格。2017年10月26日,我国药监部门联合卫生部发布相关制度文件,将药物临床试验机构由原先的资格认定制改为备案管理,并于2019年11月29日起正式实施。临床试验机构备案管理是指:开展临床试验前,申请人应当向国家药监局下属国家药品审评中心提出临床试验申请,自申请受理并缴费之日起60日内,如未收到国家药品审评中心否定或质疑意见的,可按照提交的方案开展药物临床试验。
国内临床试验研究者需要同时从事临床医疗和临床试验,医疗工作压力大,难以匹配足够的时间和精力投入临床试验项目。因此,SMO企业搭建临床试验的执行体系,通过CRC提供专业现场管理服务,协助研究者执行临床试验中非医学判断性质的事务性工作,服务内容包括:协助医生进行患者预筛,由医生进行知情同意,提高筛选入组效率;协助医生及时完整的完成数据收集和EDC录入;进行受试者预约,提示研究者工作安排,解决研究者时间分配问题;协助研究者递交伦理材料;协助进行临床研究项目的协调工作等,让传统的临床试验由“监查”推动,转化为商业化的“机构现场管理”自主推动,实现临床试验是“做出来的,不是监查出来的”高效理念和效率。
2009年-2014年期间,SMO公司数量日益增加,其临床试验项目主要来源于跨国制药企业。但是此阶段的SMO公司的经验和管理尚在摸索阶段,新药研发行业包括研究机构对SMO并未过多关注。
2015年-2018年期间,国家药监部门开展药物临床试验数据自查核查以后,行业内发现SMO参与的临床试验,其质量更能满足药政部门的核查要求。因此,不仅是国外制药企业,国内制药企业的新药临床试验对SMO服务的需求在短期迅速增加。
中金企信国际咨询公布的《2021-2027年中国临床试验现场管理外包(SMO)行业现状分析及赢利性研究预测报告》
(2)SMO行业发展现状:
①人员规模持续增长,行业处于快速发展期:随着国内临床试验机构数量的不断增长,临床试验机构分布的城市数量也随之增加。为了实现对临床试验机构的覆盖,需要SMO企业向临床试验所在城市或临近城市派驻更多CRC人员,因此CRC人员规模也是衡量SMO企业服务能力的重要指标之一。近年来国内CRC人员数量迅速增长,SMO行业处于快速发展期。
②服务周期较长:药物从研究开始到上市销售是一项高技术、高风险、高投入和长周期的复杂系统工程,以化学药为例,主要研究与开发工作包括化合物研究、临床前研究、临床试验申请与批准、临床研究、药品注册申请与审批以及上市后持续研究。
临床研究的目的是通过对新药进行广泛的人体试验,评估其对疾病治疗的有效性以及对人体的安全影响。该阶段主要分为I、II、III、IV期。新药在批准上市前,需要进行I、II、III期临床试验;新药上市后,随着大批的患者开始使用该类药物,药品生产企业应当持续考察新药的生产工艺、质量、稳定性、疗效及不良事件等情况,并定期向监管部门递交报告。根据需要,制药企业还需按照监管部门的要求开展IV期临床试验,以进一步研究药物的疗效和安全性。
临床研究时间周期可能长达10年,整个过程不可预见因素较多,因此,大
制药企业会选择将药物研发部分或全部委托给CRO公司,并且越来越多的制药企业将临床试验现场管理委托给SMO公司。
③人员区域发展不平衡进一步改善:2004年2月,国家药监局和国家卫健委共同颁布《药物临床试验机构资格认定办法(试行)》(以下简称“资格认定办法”),规定自资格认定办法颁布之日起,对原药理基地和新申报机构资格的医疗单位进行药物临床试验机构资格认定工作,自此我国第1个临床试验认定机构(GCP机构)产生。2009年5月原CFDA和原卫生部共同下发了《关于开展药物临床试验机构资格认定复核检查工作的通知》,启动机构资格认定复核检查工作,随后颁发《药物临床试验资格认定复核检查标准》(以下简称“复核标准”)认定办法,使GCP机构的自我管理得以进一步完善。
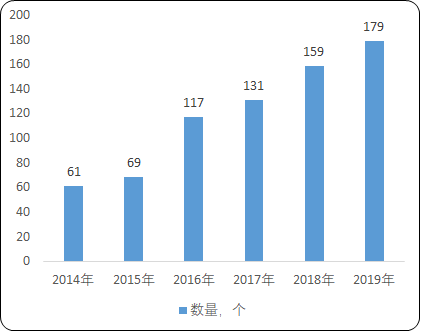
受到各地区临床试验数量的影响,全国拥有CRC人员的城市中,北京、上海、广州等一线城市人数规模占有绝对优势,反映了我国CRC人员分布区域差异化的现状。另一方面,随着新药临床试验正在向二、三线城市加快发展,近年来二、三线城市的CRC绝对数量呈明显上升趋势。根据中国CRC之家不完全统计,全国CRC人员分布城市由2014年61个城市增长至2019年179个城市,区域发展不均衡的状况正在改善。
2014-2019年中国CRC城市分布数量分析
数据统计:中金企信国际咨询
“中国CRC之家”系由医科院肿瘤医院GCP中心李树婷主任(同时兼任药物临床试验机构联盟副秘书长)发起,是DIA下设的SMO行业的学术组织,亦称“DIA中国SMO协作组”,由北京地区主要的SMO响应并辐射到全国,是中国最早、规模最大的CRC行业组织。通常由DIA发起相关活动,“中国CRC之家”负责举办与SMO、CRC有关的学术和联谊活动,增加行业交流与合作,促进行业健康发展。DIA系在美国注册的全球性、中立的协会组织,为一家国际性、非营利性、多学科的会员制协会,为医疗产品开发的专业人员提供中立、透明的论坛,推动药品和技术的合作和交流,改善全球健康。其总部位于美国首都华盛顿,在美洲、欧洲、亚洲、中东及非洲等地均有业务拓展,办事处分别位于美国宾州、瑞士巴塞尔、日本东京、中国北京和上海以及印度孟买。李树婷曾经为中国医学科学院肿瘤医院国家GCP中心办公室主任、伦理委员会秘书,目前为DIASMO协作组理事长、临床研究促进公益基金秘书长、中国抗癌协会稽查协作组指导委员等。1996年开始从事GCP和伦理委员会工作,是我国最早实施GCP的人员之一。了解并掌握国内外新药临床研究的各种规范和程序,审核各种临床研究方案1000项以上,撰写国家一类新药I期临床研究方案等多项,参与新药方案设计100余项。作为主要参与者承担了国家“九·五”至“十三·五”国家重大专项项目,2014年获得中华医学会科技进步成果一等奖、中国药学会科技一等奖、华夏医学科技一等奖、教育部科技成果一等奖等。
中国CRC之家的理事单位主要系国内SMO行业领先企业,包括但不限于普蕊斯(上海)医药科技开发股份有限公司、北京联斯达医药科技发展有限公司、杭州思默医药科技有限公司、上海津石医药科技有限公司、西斯比亚(北京)医药技术研究有限责任公司等。
④多中心临床试验带动行业发展:目前,中国临床试验正处在高速变革的阶段。《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》,重点指出“改革临床试验管理,加快上市审批速度”。在放宽临床试验中心资格认定标准,鼓励医疗机构、医学研究机构、医药高等学校开展临床试验的同时,提高了对临床试验质量的要求。意见中还提出,允许境外企业和科研机构在我国依法同步开展国际多中心临床试验,及接受境外临床试验数据。该意见的发布,为在我国开展多中心临床试验和参与国际多中心临床试验创造了前所未有的发展契机。
国际多中心临床试验(MRCT)作为一种特殊形式的多中心临床试验,是在新药开发全球化的大环境下,为减少不同国家的重复临床试验而产生的,多中心临床试验近年已成为临床试验的趋势,在我国开展的药品和器械注册临床试验越来越多地采用多中心设计。由研究者自行发起的临床试验,因重视合作交流,也越来越多地选择多中心开展。在我国开展多中心临床试验、参与MRCT越来越多的大环境下,通过设计、实施、报告三个层面入手,强化设计科学性、提高试验质量和促进报告透明化,进一步规范多中心临床试验,给SMO企业带来新的发展机遇。
⑤SMO企业的管理体系和资源愈发受到重视:项目管理能力、项目执行经验及临床试验机构覆盖能力是项目申办方选择SMO企业主要的衡量标准之一。根据尚普咨询的研究报告,临床试验的具体执行过程,大部分工作量属于非医学判断的事务性工作。临床试验项目的设计越来越复杂,项目的组织、管理拆分也越来越细。通过建立有效的项目管理体系将繁琐的事务性工作流程化、分解化、高效化,形成标准化操作流程,可使所有参与临床试验的机构的执行人员处于同一个技术质量水平上,继而推进试验进度和质量。具备完善项目管理体系的SMO企业可将标准的操作流程,快速复制到其他临床试验项目,提高临床试验项目的效率。
同时,庞大的患者管理数量,与临床试验机构稳定的合作关系,是有效满足申办方需求的必要条件。大型SMO公司通常经营时间较长,积累了丰富的数据库资源:项目执行数据库有助于协助申办方制定患者招募计划及方法,提高患者筛选成功率,加快临床试验开发进程;机构数据库利于申办方对试验场所的评估与筛选,特别是多中心试验的顺利开展。
⑥备案制赋予行业新机遇:2004年,我国药监部门联合卫生部颁发《关于印发<药物临床试验机构资格认定办法(试行)>的通知》(国食药监安[2004]44号),同时颁发了《药物临床试验机构资格认定办法(试行)》及《药物临床试验机构资格认定标准》,要求从2005年3月1日起,未通过认证的机构将不再具有承担药物临床试验的资格。2017年10月26日,原CFDA官网发布了《药物临床试验机构管理规定(征求意见稿)》,药物临床试验机构由原先的认定制转化为临床试验备案制。2019年11月29日,国家药监局联合国家卫健委发布临床试验机构管理的公告(2019年第101号),药物临床试验机构由资格认定制改为备案管理。
此外,一直以来我国临床试验的整个流程主要的管理模式为“严进宽出”,即必须经过药监部门和伦理的审批,才能开展临床试验。2018年7月27日颁发的《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号),对药物临床试验审评审批的有关事项做出调整:在我国申报药物临床试验的,自申请受理并缴费之日起60日内,申请人未收到否定或质疑意见的,可按照提交的方案开展药物临床试验。药物临床试验备案制正式实施后,新的审批流程中,临床试验在国家药监局的审批将加快,届时临床研究的数量将会进一步增加,给SMO行业带来新的机遇。
(3)行业竞争及发展前景分析:
1、目前国内SMO行业仍处于早期发展阶段,数家头部企业与多家尾部企业并存:我国SMO行业于2008年左右才开始起步,目前仍处于早期发展阶段,市场格局较为分散,呈现数家头部企业与多家尾部企业并存的竞争格局。截至2020年10月末,在“中国CRC之家”登记的SMO企业总计39家,其中CRC人员规模在1,000人以上的仅4家企业,合计占行业10.26%;其中,公司、药明津石和杭州思默的CRC人员均已超过2,000人;500-1,000人的企业有3家,100-500人的企业有14家。除此以外,多为地方性SMO企业,规模较小,CRC数量通常在100人以下。
2、预计未来随着头部企业壁垒不断巩固,SMO行业集中度将不断提升:头部SMO企业在客户资源、人才规模、机构覆盖、技术、品牌与口碑等方面建立核心壁垒,且随着经营规模的逐渐扩大,客户资源进一步丰富、人才规模进一步扩大、机构覆盖率进一步提升、技术进一步更新迭代、品牌与口碑进一步建立,前述壁垒将进一步巩固,预计SMO行业集中度将进一步提升。


